









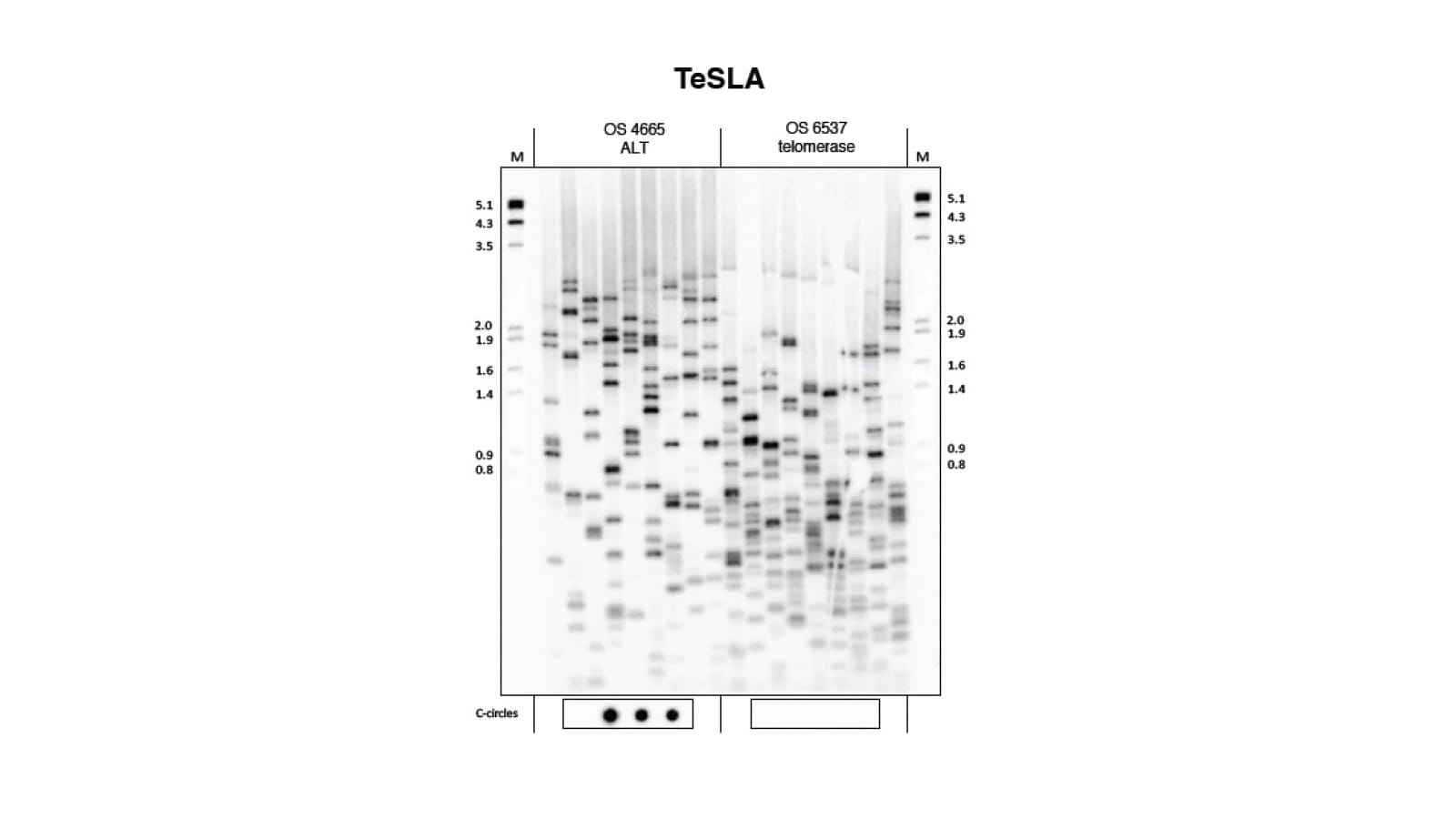
mTert induction in p21-positive cells counteracts capillary rarefaction and pulmonary emphysema.
Lipskaia L, Breau M, Cayrou C, Churikov D, Braud L, Jacquet J, Born E, Fouillade C, Curras-Alonso S, Bauwens S, Jourquin F, Fiore F, Castellano R, Josselin E, Sánchez-Ferrer C, Giovinazzo G, Lachaud C, Gilson E, Flores I, Londono-Vallejo A, Adnot S, Géli V